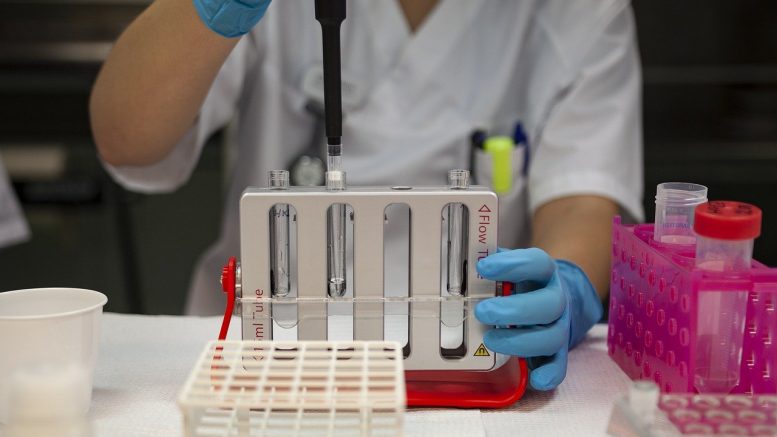
On March 17, the U.S. Food and Drug Administration (FDA) granted marketing authorization of the BioFire Respiratory Panel 2.1 (RP2.1), the first SARS-CoV-2 diagnostic test for the simultaneous qualitative detection and identification of multiple respiratory viral and bacterial nucleic acids in nasopharyngeal swabs (NPS) obtained from individuals suspected of COVID-19 and other respiratory tract infections.
The diagnostic test, which had an Emergency Use Authorization (EUA), was granted marketing authorization using the De Novo premarket review pathway, a regulatory pathway for low- to moderate-risk devices of a new type. The grant of this De Novo request marks an important step in FDA’s response to the COVID-19 pandemic because it is the first SARS-CoV-2 diagnostic test that will be permitted to be marketed beyond the public health emergency.
“Today’s action is a great demonstration of the FDA’s work to protect the public health in emergency response situations and beyond,” said FDA Acting Commissioner Janet Woodcock, M.D.
“We ensured there were tests made available quickly under EUA; and we continue to work with diagnostic manufacturers to take the next step of ensuring products are FDA reviewed for safety and effectiveness and authorized for marketing under our traditional premarket authorities.
While this is the first marketing authorization for a diagnostic test using a traditional premarket review process, we do not expect this to be the last and look forward to working with developers of medical products to move their products through our traditional review pathways.”
First SARS-CoV-2 Diagnostic Test From De Novo
The grant of the De Novo request for this test is based on additional data showing validation beyond what is needed for emergency use authorization. The FDA reviewed data from a clinical study of more than 500 test samples and a variety of analytical studies, which demonstrated a reasonable assurance that the BioFire RP2.1 was safe and effective at identification and differentiation of various respiratory viral and bacterial pathogens.
With granting of the De Novo for the BioFire RP2.1, the FDA has also revoked the EUA for this device, which was initially authorized for emergency use in May 2020. This EUA revocation and De Novo authorization do not impact the availability other tests under EUA.
“Safety, effectiveness and innovation remain important priorities for CDRH. Today’s action underscores the FDA’s ongoing commitment to expand access to testing while providing important safeguards through both our EUA authority and traditional review pathways,” said Jeff Shuren, M.D., J.D., director of the FDA’s Center for Devices and Radiological Health.
The BioFire RP2.1
The BioFire RP2.1 is for use only in individuals suspected of respiratory tract infections, including COVID-19. This diagnostic test is for the detection and identification of specific viral and bacterial nucleic acids from individuals exhibiting signs and/or symptoms of respiratory infection and aids in the diagnosis of respiratory infection if used along with other clinical and epidemiological information.
Results of the test should not be used as the sole basis for diagnosis, treatment, or other patient management decisions. Positive results of this test do not rule out coinfection with other organisms. Negative BioFire RP2.1 results in the setting of a respiratory illness may be due to infection with pathogens that are not detected by this test or lower respiratory tract infection that may not be detected by an NPS specimen.
Along with this De Novo authorization, the FDA is establishing criteria called special controls that define the requirements related to labeling and performance testing. When met, the special controls, in combination with general controls, provide a reasonable assurance of safety and effectiveness for tests of this type.
This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) pathway, whereby devices can obtain clearance by demonstrating substantial equivalence to a predicate device.


Be the first to comment on "FDA Permits Marketing of First SARS-CoV-2 Diagnostic Test"